 Western Blotting
Western Blotting
Never pray to Western Blotting God again
Yes, I know. We all have struggled with WB in one time or another. We all wanted to flush a gel or a membrane down the sink. WB is a multi-step procedure and therefore many things can and will go wrong (remember Murphy’s law, duh).
If you are new to the field, here are the major steps in WB procedure that require your attention, strategical planning and meticulous optimization:
If you want to get things right and get the blots as shown in the picture below, you MUST LEARN HOW TO FOLLOW MY VALIDATED PROTOCOL of Multi-Strip Western Blotting (MSWB), and not to try "reinventing the bicycle" or skip the steps. You can do any adjustments that you like later, i.e. when everything finally works. You can test different antibodies, their dilutions, the transfer settings etc. But only after you have an established WORKING PROTOCOL. And please, do not be afraid to go green and recycle. For instance, you can recycle cell culture media flasks: simply wash them and put under UV overnight. When I finish my blotting, I always recycle square Petri dishes that I incubate my membranes in.
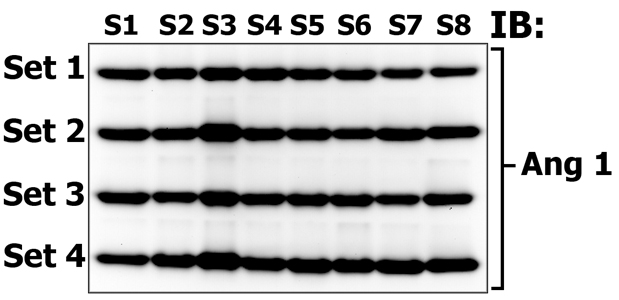
This figure demonstrates the reproducibility of sampling and MSWB procedure. Eight different animal tissue samples (S1 - S8) were made in quadruplicates and loaded onto separate gels (Set 1 through Set 4). Electrophoretically resolved proteins that migrate in 10-20 kDa gel zones in Set 1, 2, 3 and 4 accordingly, were then transfered onto one membrane, which was subsequently probed (immunoblotted, IB) with antibodies against Angiogenin 1 (14 kDa). Please note that protein transfer, membrane blocking, antibody incubation, washing and protein detection steps are performed under identical conditions despite the fact that our samples were loaded onto different gels! In this way you can quantitatively and qualitatively compare the signals of multiple protein bands. Providing that one is using the 10 well gel system, load molecular weight marker on the first and last well (for easy gel strip cutting) and load 7 gels with samples, then one can simulatenously compare the signals of 56 bands on one membrane! If you use 12 well gels - 70, an 15 well gels - 91! All major expenses come from gels, which you can cast on your own and redcue the expenses to minimum.
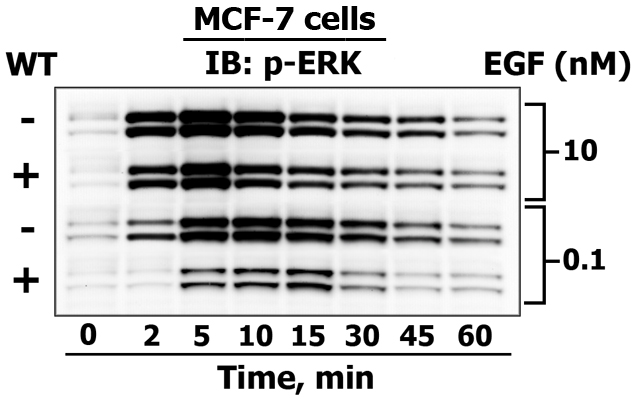
This figure demonstrates how MSWB is used to reveal the differences between 42/44 kDa ERK1/2 activating phosphorylation kinetics (from zero (reflects basal state phosphorylation level) up to to 60 minutes) in response to high and low dose of EGF treatment (10 nM and 0.1 nM) in untreated control human breast adenocarcinoma MCF-7 cells (-) and cells that were pretreated (+) with the PI3-kinase inhibitor wortmannin (WT). In addition, from the same sample loading we also detected phosphorylated Akt (60 kDa) and EGF receptor (165 kDa) levels, as well as monitored the expression of p85 PI-3-kinase subunit (85 kDa), Grb2 (25 kDa) and COX IV (17 kDa) (as in-gel loading controls). For this reason, we never do blot stripping, which is an outdated form of showing equal protein loading. If you are not convinced whether you have comparable protein levels in a gel, it is best to dye a gel with Coomasie stain.
The original protocol of Western Blotting procedure that we routinely use in my laboratory you can find here. The newer improved version of MSWB protocol is available for download here. And finally, the newest more concised version of our protocol, including the experimental design tips, can be found here. PLEASE, cite me if you find it useful, as good advice does not cost anything, right?