Growth factor-stimulated cell fractionation into soluble (cytosolic) and insoluble (crude membrane) fractions
Grow your cells in 100×20mm tissue culture dishes to confluence. For one time point I usually combine two 100×20mm plates or growth the cells in bigger 150x20 mm dishes.
Starve cells overnight in serum-free media; If your cells release significant amounts of growth factors, you may want to change the media into fresh serum-free media prior to cell stimulation (optional).
Add selected dose of growth factor into the serum-free media, rotate a dish few times and stimulate for certain period of time (e.g. 5 min) at 37ºC.
If inhibitory effects are to be evaluated, then pre-treat cells with inhibitor at least 30 minutes prior to stimulation. Leave control cells without inhibitor, add just the same amount of inhibitor solvent (e.g. DMSO).
10 seconds before stimulation time is over, start removing media by vacuum suction.
Overlay cels with 1.5 ml (for one 100×20mm plate) of ice-cold cell permeabilization buffer and leave on ice for 10 min. Some cell lines (like HEK293) will be fully permeabilized within this time, some will require slightly longer incubation time, which needs to be evaluated empirically.
Take a scraper and very gently brush all permeabilized cells into the bottom of the dish. Collect the buffer and cells into 2 mL Eppendorf tube, which should be labeled as MEM or M (for crude Membrane fraction).
Spin down the tube for 1-2 min in an ultracentrifuge at max speed (10,000×g) at +4ºC.
Transfer all supernatant into separate 2 mL Eppendorf tube, labeled as CYT or C (for cytosolic or soluble fraction). Do not leave a drop. If needed, use gel loading pipette tip to take out all the fluid. Do not disturb the pellet!
Resuspend cell pellet in 250 μL (up to 500 μL if using 150x20 mm plate) of ice-cold cell lysis buffer. Vortex. Centrifuge the extract at 10,000×g for 10 min at 4ºC to remove debris.
This will yield Triton X-100 soluble membrane fraction only (SOL-M).
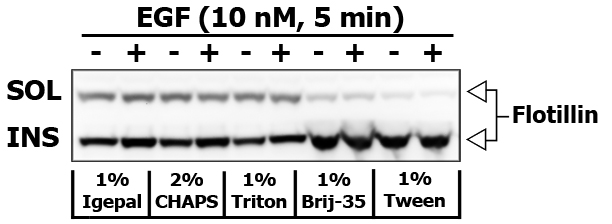
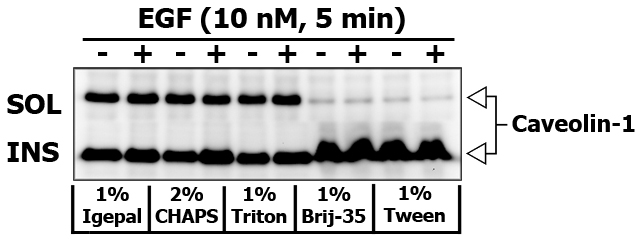
If you would like to obtain insoluble (INS-M) membrane fraction, then remove the supernatant to the seprate tube, and add 250 μL of modified RIPA cell lysis buffer or cell lysis buffer supplemented with 60-70 mM n-Octyl-beta-D-Glucoside to the pellet. Do not vortex, but pipette gently up and down to break the pellet. Centrifuge the extract at 10,000×g for 10 min at 4ºC to remove debris. Triton X-100 insoluble fraction is usually enriched in Flotillin and Caveolin.
Centrifuge all extracts at 10,000×g for 10 min at 4ºC to remove remaining debris.
Resuspend the aliquot of supernatants in Laemmli/NuPAGE LDS buffer to prepare the samples for subsequent electrophoresis.
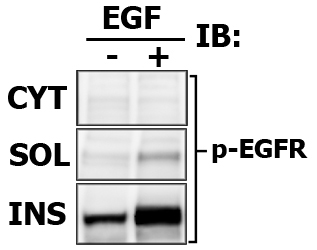
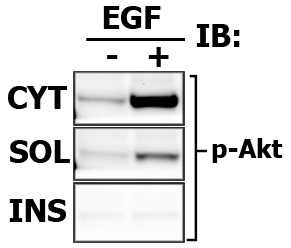
Assay cytosolic and particulate fractions for protein translocation and/or activation.
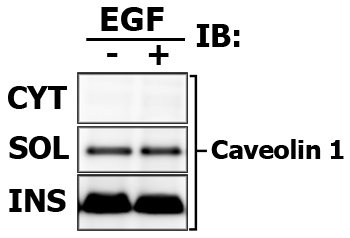
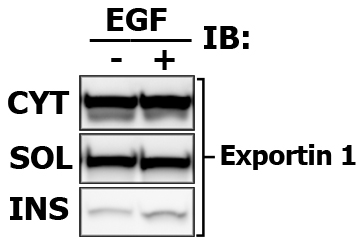
Present the results nicely! In addition to your protein of interest, detect the protein markers that are unique for each fraction. It will show that your fractions are not contaminated. For membrane fraction you may detect Ras, for cytosolic fraction - MEK or Caveolin.
Expected results indicating the relative signal intensity proportions (again, this is one of the benefits of Multi-Strip Western Blotting, and i draw the frames only to aid viewer):
Note that in this case the Cytosolic fraction (CYT) was extracted with CPB, then membrane soluble fraction (SOL) is made with in 1% Brij-35-based CLB, and finally Membrane insoluble fraction (INS) is extracted with 1% Tween-20-based modified RIPA buffer.
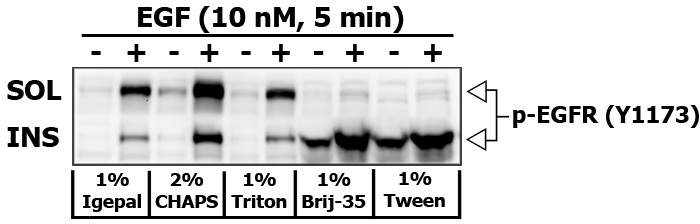
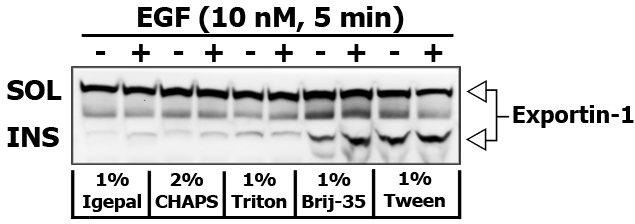
Some detergents are more selective than others, and disrupt fewer lipid–protein interactions. I did an experiment to compare the strength of different detergents (1% Igepal, 2% CHAPS, 1% Triton X-100, 1% Brij-35 and 1% Tween-20) to extract and re-extract certain proteins in 10 nM EGF stimulated PL5 cells (5 min). Brij-35 and Tween-20 are known for their inability to disrupt and dissolve the lipid rafts.
After removing the detergent soluble fraction, the pellet was resuspended in the buffer containing an appropriate detergent supplemented with 70 mM n-Octyl-beta-D-Glucoside, which solubilizes lipid rafts very well.
Here is the images of extracted proteins in soluble (SOL) versus insoluble (INS) membrane fraction.
 Cell Fractionation
Cell Fractionation